Key words
QE HF; DDA; DIA; Hela; protein identification; quantitative proteomics
introduction
Data-independent acquisition (DIA) is a new method of mass spectrometry data acquisition developed in recent years [1] . Its philosophy is to perform relative/absolute quantification of proteins with secondary fragment ions. In the DIA scan mode, ultra-high resolution mass spectrometry fragments all precursor ions in a specific mass range, collects fragment ions of all parent ions, and rapidly scans all fragment ions in the adjacent parent ion wide port in sequence. DIA's data contains retention time and intensity information for all fragment ions. The peptides can be identified and quantified by objectively extracting multiple daughter ions of the same peptide with a very small mass deviation wide mouth (eg, 10 ppm) and calculating the intensity of the daughter ions. DIA quantification has better selectivity and accurate quantification than traditional DDA based on parental ion intensity [1] , so DIA has become a new development direction of quantitative proteomics.
Q Exactive HF is a new electrostatic field orbitrap ultra-high resolution mass spectrometer introduced by Thermo Fisher Scientific at ASMS 2014 (Figure 1) [2,3] . Q Exactive HF uses Advanced Quadrupole Technology (AQT) to increase ion transmission efficiency by at least 2x; Ultra High Field Orbitrap technology improves Orbitrap scanning speed at 15000 resolution The scan speed of the graph is 20 Hz. These two technologies improve the ability of QE HF to collect DDA and DIA data. In this paper, the DDA identification ability and DIA quantitative ability of QE HF were investigated by using a 1-hour fast chromatographic gradient, and the DDA quantification and DIA quantification ability were compared from the quantitative peptide number and CV.
Experimental condition
Experimental materials and methods
Pierce HeLa Protein Digest Standard (Cat. No. 88329), diluted to 500 ng/μl, EASY-nLC injection 1 μl, 500 ng for DDA, DIA data acquisition, 3 replicates for each acquisition mode.
High performance liquid chromatography
High Performance Liquid Chromatograph: EASY-nLC 1000 (Thermo Scientific TM )
Analytical column: laboratory-made C18, 15 cm, ID 75 μm, 3 μm
Mobile phase: A: 0.1% aqueous formic acid; B: 0.1% acetonitrile formic acid gradient: 60 min, 3/0 – 6/2 – 22/48 – 40/53 – 80/55 – 80/60 (%B/min )
Flow rate: 300 nL/min
Mass Spectrometry
DDA data collection:
Mass spectrometer: Q Exactive HF (Thermo Scientific TM );
Ion source: NanoFlex ion source; ion mode: positive ion;
Spray voltage: 1.8 kV; capillary temperature: 275 ° C; S-Lens RF: 55%;
Resolution: Level 120,000@m/z 200, Level 15000@m/z 200; Level 1
AGC: 3e6, Maximum IT: 50ms;
Collision energy: NCE 27%; Fixed first mass: 110 m/z
DIA data collection:
Mass spectrometer: Q Exactive HF (Thermo Scientific TM ) ;
Ion source: NanoFlex ion source; ion mode: positive ion;
Spray voltage: 1.8 kV ; capillary temperature: 275 °C; S-Lens RF : 55% ;
Target m/z window: 400–1000 ; isolation window: 12Da ;
Collision energy: 27% ; fixed first mass: 200 m/z ; AGC target: 1e6 ;
Maximum ion injection time: atuo ; loop count: 50
data processing
Proteome Discoverer protein identification process: human protein database ( uniprot human_201309 ), parent ion mass deviation : 10 ppm ; fragment ion mass deviation: 0.02 Da ; fixed modification: cysteine ​​alkylation ( +57.021 Da ); dynamic modification: A Oxidation of methionine ( +15.995 Da ); deamination of asparagine and glutamine ( +0.984 Da ); enzyme: trypsin ; miss cleavage site: 2 ; FDR< 0.01
Skyline DDA, DIA protein excretion process: DDA quantitative skyline MS1 filtering function, extraction of segments for the highest intensity of each peptide three parent ion isotope peaks, idotp ≥ 0.8; DIA quantitative skyline DIA function provided 12 Da isolation window Peak extraction was performed on the 5 daughter ions with the highest intensity of the peptide , mProphet was scored, and FDR < 0.01 was controlled .
Experimental result
1. DIA data collection and analysis process
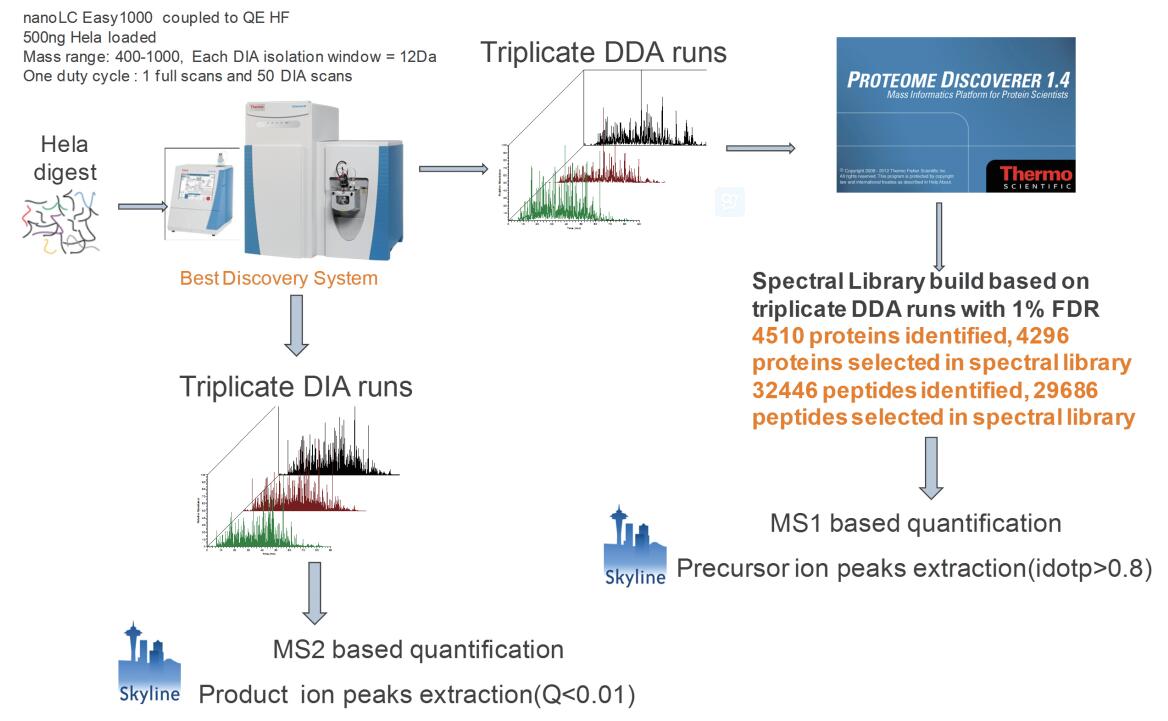
Figure 1. DDA quantification and DIA quantification protocol
500 ng Hela cell lysates was performed three times 1 h DDA gradient analysis, Proteome Discoverer 1.4 database search. The identified protein and peptide information was imported into the skyline as candidate quantitative proteins and peptides. Based on the parent ion quantification , the DDA data was extracted by the MS1 filtering function in the skyline . 500 ng Hela cell lysate was subjected to 3 target m/z windows 400–1000 , and the DIA analysis of the isolation window 12 Da was performed . Based on the quantification of secondary ion, peak extraction was performed on the daughter ion using the skyline DIA function, mProphet was scored, and the peptide with Q value < 0.01 was selected as the quantitative peptide.
Before analyzing the DIA data, it is necessary to establish a spectral library. The spectrum library contains the peptides identified by all proteins in the mass spectrum, as well as the retention time of the peptides, the fragment ion mass-to-charge ratio, and the fragment ion intensity. Data-dependent scanning is the best way to build a data collection method for a spectral library. Three hundred DDA data were collected from 500 ng Hela cell lysate . Raw data was retrieved by Proteome Discoverer and controlled for FDR < 1% . The three DDA identification results were combined and imported into the skyline to create a spectrum library. In addition to giving identification information for proteins and peptides, DDA data can also be quantified based on the intensity / peak area of ​​the parent ion . Skyline MS1 filtering function in a plurality of parent ions isotope peaks were extracted, and scoring (idotp value) [4] The isotope distribution. Idotp represents the similarity between the measured isotope distribution and the theoretical isotope distribution. A parent ion having an idotp value greater than 0.8 was screened as a reliable quantitative peptide.
500 ng Hela cell lysate with the same column, the same gradient chromatography, the QE HF switches to scan mode DIA DIA data acquisition was performed three times. In one scanning cycle, the target ratio of the parent ion mass range 400-1000 nuclear quadrupole isolation window 12 Da, 50 comprising MS / MS scan. Each MS/MS spectrum contains fragment ion information for all parent ions within a 12 Da window. The QEHF uses an ultra-high field Orbitrap with a scan speed of 20 Hz , so each cycle takes about 2.4–3 s and is compatible with the chromatogram (Figure 2 ). When Skyline processes DIA data, the highest intensity fragment ions are selected from the spectrum library for peak extraction. Skyline embedded software mProphet scored according to feature a plurality of peaks of ions of the same peptide. Feature includes sub-ion co-elution peak shape, retention time deviation, dotp value, signal to noise ratio, and the like. In order to distinguish the false positive sub-ion peaks, mProphet can create a decoy library, or you can use the second-ranked peptide as decoy to calculate FDR [5,6] . Peptide segments with FDR < 0.01 were screened as authentic quantitative peptides (Figure 1 ).
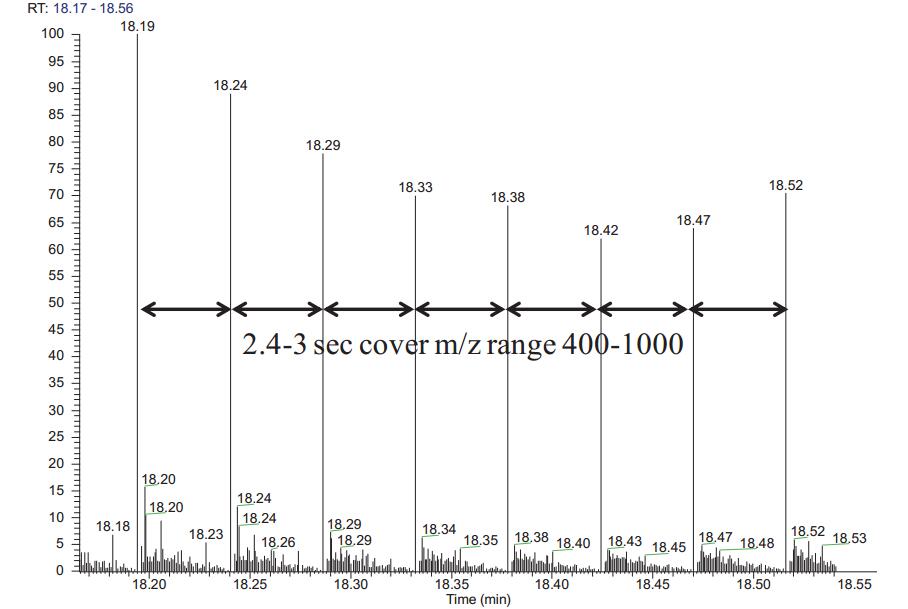
FIG 2. QE HF acquired nuclear scan target substance 50 MS / MS than the desired time window 400-1000 2.4-3 s
2. DDA identification results and spectrum library creation
500 ng Hela cells were subjected to DDA data acquisition with a 60 min chromatographic gradient . Within 60 min, triplicate, QE HF were collected into 49065, 49031, 48885 spectra identified 22030, 21820, 21654 peptides corresponding to about 3909, 3900, 3878 proteins. The peptides and proteins were combined three times, and a total of 32,446 peptides and 4510 proteins were identified (Fig. 3 ). Protein and peptide identification results were imported into the skyline to create a spectral library. Through the library, the candidate peptides and proteins to be quantified are established in the skyline . In order to improve the accuracy of protein quantification, some peptide restriction conditions are set in the skyline , m/z 400–1000 , parent ion charge 2 + – 4 + , no missing cleavage site. 32446 peptide in peptide meet these conditions are 29686, 4296 correspond to proteins. DDA in quantitative experiments, adding three highest intensity peaks for each candidate peptide isotope segment (M, M + 1, M + 2) as a quantitative ion, ions were generated quantitative 89,027. In the DIA quantitative experiment, five of the highest intensity product ions ( b 3 -b n , y 3 -y n ) were added for each peptide segment , and a total of 145,736 quantitative ions were generated .
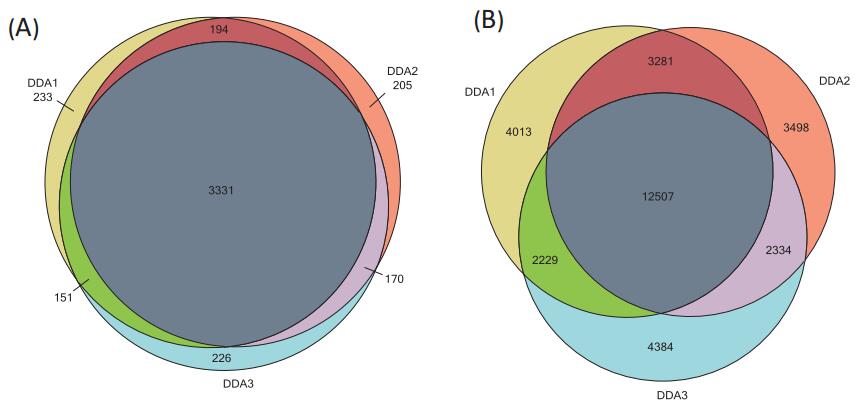
FIG 3. 500 ng Hela cell lysate results identified three times with DDA (A) of the identified protein cross-cap (B) of FIG cross cover the peptides identified
3. Comparison of DDA quantification based on parent ion and DIA quantitative results based on daughter ion
For DDA quantification, skyline will extract the parent ion isotope peak from the DDA data first-order spectrum. The peptides with well-distributed isotope distribution and theoretical isotope distribution were screened as reliable quantitative peptides, ie idotp > 0.8 . Since the same chromatographic gradient was used in the DDA and DIA experiments, the retention time information of the peptides in the DDA experiment can be passed to the DIA experiment. Therefore, in the DIA quantification, the skyline is limited to extract only the product ions in the peptide library for 5 min . The retention time limit can reduce the complexity of DIA data processing and increase the accuracy of quantification. At the same time , the most reliable quantitative peptides of the peptide with Q value < 0.01 were screened .
Three DDA experiments were performed to quantify 25214 , 25515 , and 24873 peptides from the first-order spectrum ; 3969 , 3975 , and 3917 proteins; the quantified peptides accounted for 84% of the total candidate peptides ; the CV value of the peak area was 20 % or less of 58.81% of the total quantitative peptides; the CV value representing 25.25% (FIG. 4) at 10% or less. Three DIA quantifications were quantified to 27558 , 27604 , and 27483 peptides, respectively; 4067 , 4080 , and 4073 proteins. 93% to quantitative Zhang candidate peptides peptides; the CV value of the peak area at 20% or less of 90.3% of the total quantitative peptides; the CV value representing 69.01% (FIG. 5) at 10% or less. It can be seen that DIA is able to quantify more peptides, and the peak area CV value of the quantitative peptide is much smaller than DDA .
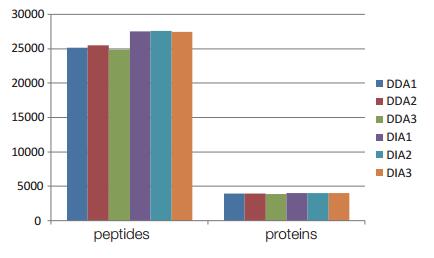
Figure 4. Experiment 3 and 3 DIA DDA experiments to quantify peptides and proteins
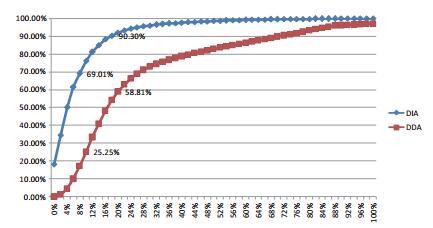
Figure 5. DDA quantification and DIA quantification of parent and daughter ion peak areas CV
in conclusion
DDA is generally used for protein / peptide identification and can be quantified based on parent ions. In very complex matrices, it is very susceptible to interference by other peptides, resulting in inaccurate quantification and excessive CV values. DIA quantification is based on the quantification of secondary ion ions. The quantification of sub-ions increases the selectivity of quantification and reduces the interference of other peptides. The quantification is more accurate than the quantification based on the first-order peak area, and the CV value is small. In this experiment, DDA quantification and DIA quantification ability were evaluated using 500 ng of Hela cell lysate . DDA experiments identified three peptides 32446, 4510 proteins. The identified proteins and peptides were imported into the skyline to establish a spectral library, and 4296 candidate quantitative proteins and 29686 candidate quantitative peptides were screened as the common quantitative target of DDA and DIA . DDA quantified 84% of the peptide, 92% of the protein; while DIA quantified 93% of the peptide, 95% of the protein. DIA more than DDA quantitatively to peptides and proteins, while the CV DIA quantitative ions is much smaller than the peak area of the CV DDA quantitative parent ion peak area. The results indicate that the quantification based on the secondary ion is due to the quantification based on the primary parent ion.
QE HF can identify and quantify 4000 proteins from 500 ng Hela cell lysate in a one-hour gradient , thanks to better ion transport efficiency and ultra-fast scanning speed of QE HF . At present, DIA quantitative analysis based on one-dimensional reversed-mass spectrometry is not suitable for two-dimensional chromatography-mass spectrometry. However, it is possible to increase the separation efficiency to achieve full coverage of the cellular proteome by extending the gradient of the one-dimensional reversed phase chromatography. In 2014 , Matthais Mann's team used a one-dimensional reversed-phase chromatogram gradient of mouse NSC-34 and N2a cell lysates for 240 min to identify more than 8,000 proteins, which basically reached the full coverage of the cell line protein group [7] . The identification of peptide proteins by DDA and the establishment of a spectral library, DIA protein quantification may become a new development direction of quantitative proteomics.
references
1. Gillet, L C. et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept forconsistent and accurate proteome analysis. Mol Cell Proteomics, 2012, 11(6): O111 016717.
2. Kelstrup, C D. et al. Rapid and deep proteomes by faster sequencing on a benchtop quadrupole ultra-high-field orbitrap mass spectrometer. J Proteome Res, 2014, 13(12): 6187-6195.
3. Scheltema R A. et al. The Q Exactive HF, a benchtop massspectrometer with a pre-filter, high performance quadrupole andan ultra-high field orbitrap analyzer. Mol Cell Proteomics, 2014,
13(12): 3698-3708
4. Abbatiello S E. et al. Design, implementation, and multi-site evaluation of a system suitability protocol for the quantitative assessment of instrument performance in LC-MRM-MS. Mol Cell Proteomics, 2013, 12(9):2623- 2639
5. Reiter, L. et al. mProphet: automated data processing and statistical validation for large-scale SRM experiments. Nat. Methods.2011 , 8 (5): 430-435
6. Röst H L. et al. OpenSWATH capable automated, targeted analysis of data-independent acquisition MS-data. Nat. Biotechnol. 2014,32(3):219-223
7. Horburg D. et al. Deep Proteomic Evaluation of Primary and Cell Line Motoneuron Disease Models Delineates Major Differences in Neuronal Characteristics. Mol Cell Proteomics, 2014, 13(12):3410-3420
Organic Fructo-Oligosachharide Powder 95%
Organic Fructooligosaccharides,Fructooligosaccharides Baby Formula,Fructooligosaccharide Diabetes,Fructooligosaccharide Powder,Fructooligosaccharide Diet
Qingdao Bailong Huichuang Bio-tech Co., Ltd. , https://www.qdblcycn.com